

ERC and DFG Emmy-Noether Group “Statistical Methods in Phylogenetics and Macroevolution”
ERC and DFG Emmy-Noether Group “Statistical Methods in Phylogenetics and Macroevolution”
(Prof. Dr. Sebastian Höhna)

Our research group works on a wide range of statistical methods, ranging from evolutionary biology, population genetics and phylogenetics to paleo-phylogenetics. Our primary aim is to develop new statistical models and computational methods to answer one of the major questions in evolutionary biology and paleobiology: what are the causes and processes that have shaped biodiversity over time and space? We want to understand why some groups of organisms, like cichlid fishes, contain thousands of species, while others, like crocodiles, have very few.
The group is primarily funded by an ERC Starting Grant, a DFG Emmy Noether Project and some specific DFG research grants (e.g., within the two priority programs TaxonOMICS and G-Evol). Our main goal is to develop statistical methods to study the biological processes that produce and impact current-day biodiversity. We are taking a phylogenetic approach to describe the relationship among species (both extant and extinct) with a specific focus on the divergence time between species. Ultimately, we want to study what processes drive historical biodiversity and are responsible for the fluctuations, such as major increases and decreases, of biodiversity over geological timescales. To answer these questions, we will take a multidisciplinary approach combining statistics, computer science, paleobiology, and evolutionary biology.
MacDrive (ERC-funded project)
The MacDrive project pursues a holistic approach to understanding the drivers of diversification by integrating fossil taxa into phylogenies of extant taxa. The project aims to address five key questions:
- Are diversification rate estimates from phylogenies without fossils reliable?
- What patterns of diversification rates can be inferred from phylogenies?
- How have diversification rates changed over time and among lineages for major groups such as Artiodactyla, Carnivora, Crocodyliformes and Squaliformes?
- Are diversification rates correlated with environmental factors and/or species-specific factors?
- Are species-specific factors correlated with survival probabilities of mass extinction events?
To answer these questions, the project involves developing new statistical models, implementing them in open-source software with efficient algorithms, producing new morphological datasets and phylogenies, and performing large-scale statistical analyses about macroevolutionary diversification.
A key component of this project involves how to place extinct taxa represented by fossils into evolutionary trees, from modeling how morphological characters evolve to using the FBD (Fossilized-Birth-Death) class of models to reconstruct how lineages have speciated, gone extinct, and have been sampled in the fossil record.
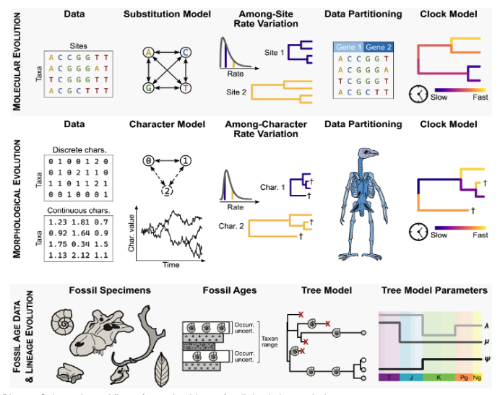
Schematic workflow of a total-evidence fossil tip-dating analysis. For more details, see Heckeberg et al. 2025.
To test for lineage-specific shifts in diversification rates, we developed the PESTO program (Phylogenetic Estimation of Shifts in the Tempo of Origination). PESTO can estimate branch-specific diversification rates and branch-specific rate shift events efficiently even for species-rich phylogenies (> 20,000 taxa). PESTO uses an existing model, the birth-death-shift model, but uses a novel algorithm based on belief propagation techniques to calculate branch rates.
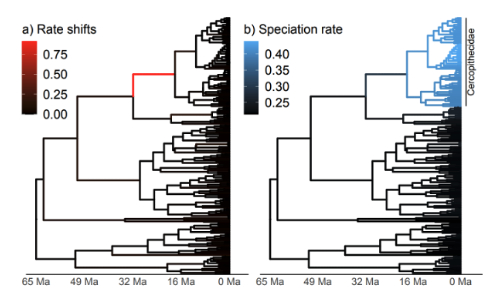
Example of a lineage-specific diversification rate analysis for primates. On the left we show the branches with supported rate shift events, and on the right we show the lineage-specific speciation rate (Kopperud & Höhna 2025).
MacDrive-Trait: Studying macroevolutionary drivers of trait evolution
On a macroevolutionary scale, i.e., over millions of years and across hundreds of closely related species, we can study adaptation by how the mean trait value per species evolves around a trait optimum. Over macroevolutionary timescales, this trait optimum will not be stable but instead vary over time, e.g., due to changes in the global environment, and across species, e.g., specialization on different diets. The currently most sophisticated approach uses the Ornstein-Uhlenbeck process, which contains three parameters: the rate of adaptation, the rate of drift, and the trait optimum. In this project we develop a novel approach to study trait adaptation over macroevolutionary timescales by extending the basic theory of Ornstein-Uhlenbeck processes. Specifically, we will develop a state-dependent Ornstein-Uhlenbeck model where all three parameters (the rate of adaptation, the rate of drift, and the trait optimum) can vary among species and/or vary over time.
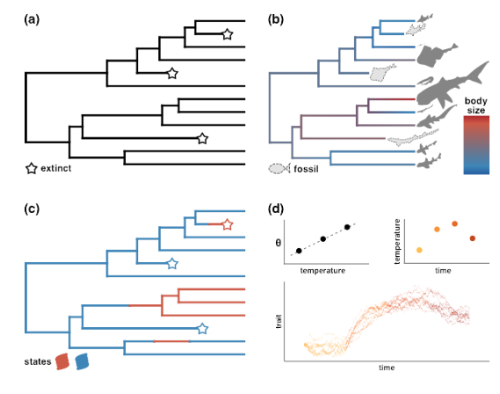
Schematic of a discrete-trait dependent continuous trait evolution. The different sub-figures depict how the body size depends on some underlying binary state.
GEvol project: Modeling gene expression evolution in fireflies
Gene expression is a key driver of trait variation, particularly among closely related species. This project aims to develop innovative methods to study gene expression evolution using phylogenetic models such as Brownian motion and Ornstein-Uhlenbeck processes, which correspond to different evolutionary scenarios. Specifically, we extend the models to allow for within-species variance, a critical yet underexplored aspect of gene expression.
In this project, we use the firefly family (Lampyridae) as a novel study system to investigate gene expression evolution. Fireflies exhibit recurrent sexual dimorphism across the phylogeny. As sexual dimorphism is inherently linked to sex-biased gene expression, this makes fireflies an ideal model for sex-biased gene expression evolution. We collect over 20 firefly species from Europe and Neotropics, with varying levels of sex dimorphism, and obtain gene expression data from both sexes. Using our newly extended phylogenetic models, we aim to uncover how sex-biased gene expression evolves and its relationship with sexual dimorphism across the firefly phylogeny.
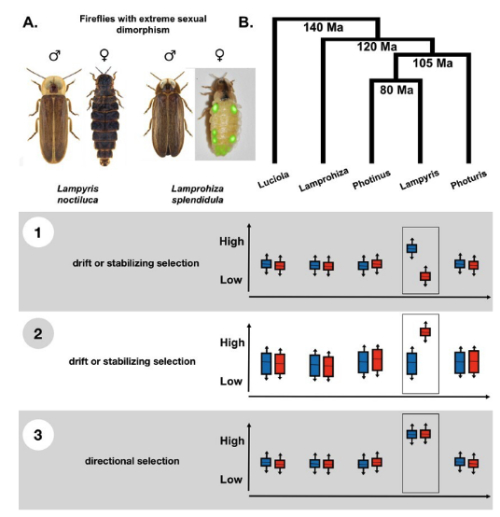
Schematic of different gene expression evolution scenarios with specific focus on sex-biased gene expression evolution. Our work here focuses on fireflies and is part of the DFG-SPP 2349.
Contact: hoehna@lmu.de